Si-Yan Chen1,
Kuang Zheng2 ![]() ,
Zhi-quan Wang3
,
Zhi-quan Wang3
For correspondence:- Kuang Zheng Email: zhengkuangzk@hotmail.com Tel:+8657755579351
Received: 28 September 2015 Accepted: 6 January 2016 Published: 28 February 2016
Citation: Chen S, Zheng K, Wang Z. Neuroprotective effects of ellagic acid on neonatal hypoxic brain injury via inhibition of inflammatory mediators and down-regulation of JNK/p38 MAPK activation. Trop J Pharm Res 2016; 15(2):241-251 doi: 10.4314/tjpr.v15i2.4
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To investigate if ellagic acid exerts neuroprotective effects in hypoxic-ischemic (HI) brain injury by inhibiting apoptosis and inflammatory responses.
Methods: Separate groups of rat pups from post-natal day 4 (D4) were administered with ellagic acid (10, 20 or 40 mg/kg body weight) orally till post-natal day 10 (D10). On D10, the rats were subjected to HI brain injury. Following HI injury, infarct size, weight and volume of the brain were measured. Apoptosis was assessed by Fluoro-Jade C staining. ex
Results: Ellagic acidmarkedly (p < 0.05) reduced infarct size, volume and tissue loss. Significant (p < 0.05) reduction in neuroapoptosis was observed on pre-treatment with ellagic acid. ex
Conclusion: Ellagic acid affords neuroprotection in HI brain injury by inhibiting apoptosis, inflammatory responses and modulating the proteins of apoptotic and MAPK pathways. Thus, ellagic acid may be a potent candidate for the treatment of HI injury.
Introduction
Neonatal hypoxic-ischemic (HI) brain injury is a major cause of perinatal mortality and morbidity [1]. Perinatal HI has an incidence rate of 18 cases in every 1000 births, affecting approximately 60 % of pre-term infants [2]. HI brain injury arising in neonatal and perinatal stages damages brain cell development, resulting in long-lasting neurologic sequelae with cerebral palsy, epilepsy, learning disabilities and motor disability as complications [3]. Brain injury in perinatal stage has multifaceted aetiology involving inflammation, a major cause in the progression of HI-induced injury in neonates and in adults [4]. Inflammation is involved in sensitizing the neonatal brain to ischaemia through pro-inflammatory cytokines which are found in increased levels [5].
Mitogen activated protein kinases (MAPKs) - p38s, extracellular signal-regulated kinases (ERKs) and c-Jun NH2-terminal kinases (JNKs) are activated in mammalian cells in response to various growth factors, cytokines and agents that cause cell damage [6]. The activated MAPKs principally function as mediators of cellular stress by phosphorylating various transcription factors, cytosolic proteins and intracellular enzymes that are chiefly involved in cell survival, apoptosis and in expression of inflammatory mediators [7].
Recently much of research work is focusing on plant-derived compounds for their neuroprotective effects. Ellagic acid is found in numerous plants, predominantly in fruits and nuts such as cranberries, pomegranates, raspberry, strawberries and walnuts [8]. Previous researchers have reported that ellagic acid possesses various pharmacological properties through regulation of several pathways such as glycogen synthase kinase-3 beta (GSK-3β)/ phosphoinositide 3-kinase (PI3-K) signaling [9]; regulation of cytokines [10]; activating antioxidant response [11]; and modulating the cell-cycle/cell survival genes [12]. Considering these biological effects the present study aimed to explore the influence of ellagic acid in neonatal HI brain injury.
Methods
Chemicals and reagents
Ellagic acid was purchased from Sigma-Aldrich, St.Louis, MO, USA. Antibodies against caspase-3, caspase-8, caspase-9, Bcl-xL, Bcl-2, Bax, Bad (Cell Signalling Technology, Danvers, MA, USA). β-actin, JNK, phospho-JNK, ERK1/2, phospho-ERK1/2, p38, phospho-p38, NF-κB(p65), p-IKBα (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used in expression analysis. All other chemicals and reagents were obtained from Sigma-Aldrich, St.Louis, MO, USA, unless otherwise specified.
Experimental animals
Pregnant Sprague Dawley rats (Guangdong Medical Laboratory Animal Co., China) used in this study was maintained on 12 h light/dark cycle with room temperature at 22 ± 1 °C. The rats had ad libitum access to water and feed and were housed individually in separate cages and monitored closely for the day of litter of pups. The day of litter was noted as postnatal day 0 (Day0). The pups were kept in cages with their littermates in conditions similar to their dams. From day 4, separate groups of pups were administered with ellagic acid (10, 20 or 40 mg/kg b.wt, orally). On D10, one hour after treatment with ellagic acid, the pups were experimentally subjected to HI. Pups not treated with ellagic acid and not subjected to HI served as control, while pups exposed to HI injury and not treated with ellagic acid served as the HI control. For animal studies, ethical approval was obtained from the ethical committee of Wen Zhou Medical college (Approval Reference Number - 4208211) and was performed in compliance with the guide for care and use of laboratory animals [13].
Hypoxic-ischemic (HI) brain injury procedure
Ten-day-old pups were subjected to HI brain injury as described by Shen et al [14]. The left common carotid artery was ligated under anesthesia with 3 % isoflurane. Following recovery period of 90 min, the pups were placed in a humidified chamber containing atmosphere of 8 % O2/92 % N2 for a period of 2.5 h and were then returned to their respective cages.
Measurement of brain infarction
Following 24 h of HI injury, the rat pups were sacrificed by decapitation and brain tissues were immediately dissected out and 1 mm coronal slices were immersed in a PBS solution containing 2 % triphenyltetrazoliumchloride (TTC) at 37 °C for 25 min. The live/dead cells were determined based on the uptake of TTC. Live cells take up TTC where it is converted to red colour in live mitochondria. The viable tissues appear brick-red, and non-viable/infarcted tissue can be identified by the absence of red colour (white). The tissues were further analysed for infarct size and volume using NIH Image J imaging applications as described by Williams et al [15].
Histological analysis by cresyl violet staining
Histologic studies were performed 48 h after HI insult. The brain tissues were fixed in 4 % paraformaldehyde and embedded in paraffin. Sections (4-5 μm) were mounted on glass cover slides and were deparaffinized and rinsed with distilled water and incubated for 15 min in 1 % cresyl violet solution. After incubation, the sections were rinsed in water and destained with 1 % acetic acid solution and finally dehydrated by rinsing with ethanol. The slides were rinsed thoroughly and observed under microscopy, photographed and analysed [16].
Evaluation of neuro-apoptosis
Neuro-apoptosis was evaluated by immuno-histochemical staining for activated caspase-3 and Fluoro-Jade C staining. The analysis was performed as described previously [17]. The brain tissue sections were embedded in paraffin (5 µm) and incubated overnight at 4 °C with anti-cleaved caspase-3 primary antibody (1:200; monoclonal antibody, Cell Signalling Technology, Beverly, MA, USA). The tissue sections were further incubated with secondary antibody (1:200, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 40 min. The sections were then incubated with avidin-biotinylated peroxidase complex (Vectostain ABC-Kit, Vector Lab, Burlingame, CA, USA) for 40 min and stained with diaminobenzidine (DAB, Vector Laboratories, Burlingame, CA, USA). The immune-positive cells in the various sections of the brain tissue - hippocampal CA1, CA3 and dentate gyrus (DG) were analyzed using NIS-Elements BR imaging processing and analysis software (Nikon Corporation, Japan).
For fluoro-Jade C staining, the tissue sections of 60 µm thickness were stained with Fluoro-Jade C, a marker very specific for neurodegeneration. The apoptotic cell counts were recorded as Fluoro-Jade positive cells using Nikon Eclipse 80i microscope under 20 x magnification.
Western blot analysis
The cytosolic and nuclear fractions were prepared and subjected to western blotting [18]. The samples were homogenized in lysis buffer(5 M NaCl, ,10 % Nonidet P-40, 1 M HEPES, 0.1 M EGTA, 0.5 M EDTA, 0.1 M phenylmethyl-sulfonyl fluoride, 1 M sodium fluoride, 0.2 M sodium orthovanadate, 2 µg/mL aprotinin, and 2 µg/mL leupeptin) and centrifuged at 20,000 g for 15 min at 4 °C. The supernatant was collected and protein content was determined using Beyotime protein assay kit (Beyotime Institute of Biotechnology, Nanjing, China). Equal amounts of protein (40 µg) were separated by electrophoresis on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (10 %) and then transferred to polyvinylidene fluoride membrane (Millipore, Billerica, USA). The membrane was blocked with 5 % non-fat dry milk in tris-buffered saline containing 0.05 % Tween-20 (TBST) buffer and incubated overnight at 4 °C with primary antibodies. Membranes were washed thrice with TBST buffer and further incubated with secondary antibodies coupled to horseradish peroxidase (1:1000 dilution, Santa Cruz Biotechnology) for 2 h at room temperature. The immunoreactive bands were detected by enhanced chemiluminescence system according to the manufacturer’s protocol (Millipore, Billerica, USA). The signals were analysed by scanning densitometry (Quantity One Software Bio-Rad Laboratories, Hercules, CA, USA). The expressed protein levels were normalized to that of β-actin.
RT-PCR analysis
Expression of the genes coding for inflammatory mediators in the cortex at 24 h following H/I induction was analyzed. The mRNA levels of TNF-α, iNOS, COX-2, IL-1α, IL-1β and IL-6 and were determined. Total RNA was isolated from lysed cells using the RNeasy kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). RT-PCR was performed as described by Shen et al [14]. Total RNA was amplified by PCR using the following primers- iNOS forward-GTGCTAATGCGGAAGGTCATG, reverse-GCTTCCGACTTTCCTGTCTCAGTA; COX-2- forward-TGTCCCTTTGCCTCTTTCAAT, reverse-GAGGCACTTGCGTTGATGGT; IL-1α- forward-AAGACAAGCCTGTGTTGCTGAAGG and TCCCAGAAGAAAATGAGGTCGGTC; IL-1β-forward –CACCTCTCAAGCAGAGCACAG, reverse- GGGTTCCATGGTGAAGTCAAC; IL-6-forward- TCCTACCCCAACTTCCAATGCTC, reverse- TTGGATGGTCTTGGTCCTTAGCC; TNF-α-forward-AAATGGGCTCCCTCTCATCAG TTC and, reverse- TCTGCTTGGTGGTTTGCTA CGAC and GAPDH forward –CCAGCCTCGT CTCATAGACA and reverse-GTAACCAGGCGT CCGATACG.
Statistical analysis
The results are presented as mean ± SD (n = 3 or 6). Data were analysed by one-way analysis of variance (ANOVA) followed by post-hoc analysis using Duncan’s Multiple Range Test. The data obtained were analysed using SPSS software (version 22.0).Differences were considered statistically significant at p < 0.05.
Results
Ellagic acid pretreatment reduced brain infarct
Extensive infarction was seen in the cerebral cortical and sub-cortical areas in rats subjected to HI (). Rats pre-treated with ellagic acid (10, 20 or 40 mg/kg) had significantly (p < 0.05) reduced infarct volumes in comparison to HI control. Further a striking rise in infract weight was observed in HI induced rats pups. Ellagic acid treatment remarkably reduced the infarct weight. 40 mg dose brought more significant reductions than lower doses, suggesting dose-dependent reduction in infarct weight and volume.
Effect of ellagic acid on brain tissue histology following HI
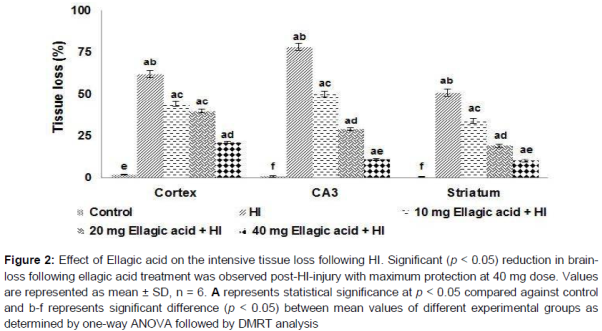
Cresyl violet staining revealed the morphology of normal viable cells as round and pale stained nuclei throughout the tissue sections of cerebral cortex, hippocampus and striatum (). Higher numbers of cells under apoptosis were observed as shrunken cells with pyknotic nuclei. Further, large areas of tissue loss were also observed in the ipsilateral cerebral cortex, striatum and hippocampus of HI control animals. Ellagic acid treatment decreased the apoptotic cell counts and also reduced the extent of tissue damage. Hippocampus had more injury when compared to the cortex and striatum. While 10 mg ellagic acid did reduce the degree of brain injury, 20 and 40 mg doses were more effective. Ellagic acid (40 mg) reduced the tissue loss to nearly 13.1 % in the hippocampus as against 78.51 % in pups that are not treated.
Ellagic acid markedly inhibits neuronal apoptosis following HI-induced brain injury
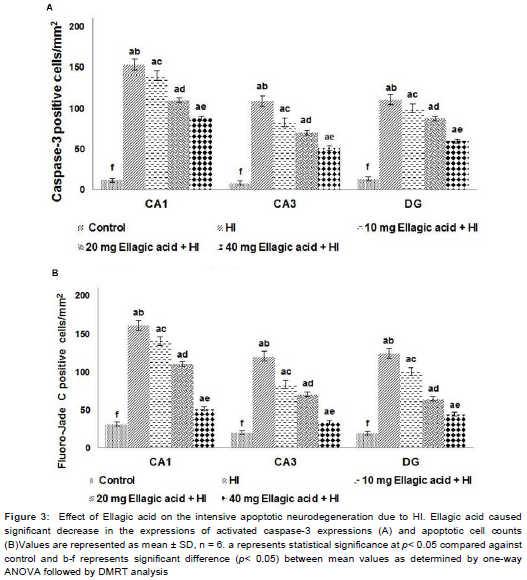
Activation of caspase-3 was considered as a measure of apoptosis. Caspase-3 positive cells were detected in CA1 and CA3 areas of hippocampus and in the DG. HI resulted in a striking (p < 0.05) increase in number of caspase-3 positive cell counts (). The expression levels of caspase-3 as determined by western blot analysis indicated significant increase (p < 0.05) following HI-injury (). Ellagic acid (10, 20 or 40 mg) pre-treatment significantly (p < 0.05) inhibited caspase-3 activation as compared to HI control pups. Immuno-reactivity towards cleaved caspase-3 in the brain tissues exhibited a dramatically reduced number of caspase-3-positive cells in the hippocampus of animals administered ellagic acid. The decrease in the expression of caspase-3 and in the number of apoptotic cell counts was observed to be dose-dependent. Furthermore, increased expression level of caspases-8 and 9 in HI was significantly reduced on ellagic acid treatment (). Also the Fluoro-Jade C positive cells (p < 0.03) were found to be lowered on ellagic acid pre-treatment ().
Effect of ellagic acid on apoptotic pathway proteins
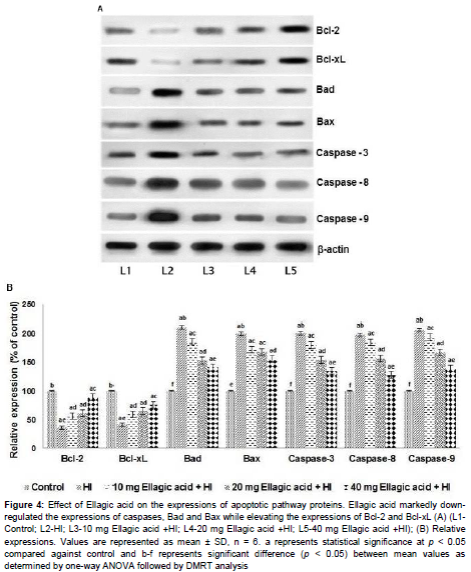
The expression of pro-apoptotic (Bad and Bax) and anti-apoptotic proteins (Bcl-2 and Bcl-xL) were assessed following HI insult. Bcl-2 family proteins are important regulators of apoptosis. In our study, HI-insult resulted in multi-fold increase in expression of Bad and Bax proteins with significant decrease (p < 0.05) in Bcl-2 and Bcl-xL expressions. Ellagic acid administration caused a tremendous decline in the expression levels of pro-apoptotic proteins while it enhanced survival proteins- Bcl-xL and Bcl-2 (). The expression of proteins were in line with the observed caspase-3 expression and apoptotic cell counts.
Ellagic acid modulated the expression of MAPK proteins
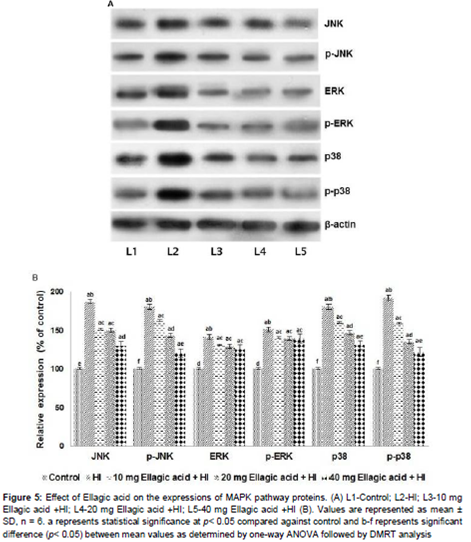
The results reveal a significantly (p < 0.05) raised levels of JNK, p-JNK, p-38 and p-p-38. Not much rise in ERK1/2 expressions were observed. Ellagic acid strikingly down-regulated the expressions of JNK and p38MAPK (). While significant suppression of JNK was observed, ellagic acid brought about only a small decrease in the expression of ERK1/2. The inhibitory effects of ellagic acid observed were dose-dependent.
Influence of ellagic acid on expression of inflammatory mediators
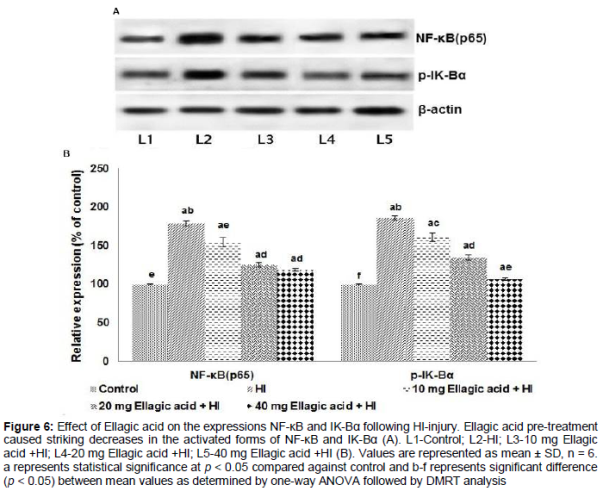
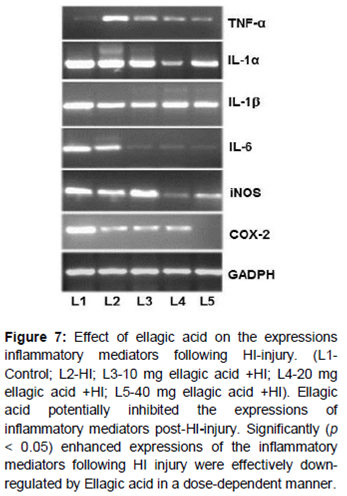
A striking increase in the expression of p65 NF-κB and p-IK-Bα on HI-injury was observed. Interestingly, ellagic acid administration caused a remarkable decrease in NF-κB and p-IK-Bα () and also in the mRNA levels of TNF-α, COX-2, IL-1α, IL-6, IL-1β and iNOS ().
Discussion
Perinatal HI brain injury is a common cause of mortality and neurological deficits in infants and children [19]. The pathological process that may possibly contribute to neuronal death and dysfunction includes inflammatory responses, apoptosis, excitotoxicity and oxidative stress [20]. Unfortunately, the therapeutic interventions are limited.
In this study, ellagic acid (10, 20 or 40 mg/kg) administered for a week significantly protected the neonatal brain from HI injury. The infarct size and volume were reduced. Studies have reported that neonatal brain cells exhibit features of apoptosis following HI insult [21].Apoptosis has been reported as a typical mechanism of cell death in the developing brain. Cresyl violet and Fluoro-Jade C staining revealed enhanced apoptosis that was significantly reduced by ellagic acid. Further, it has been reported that both caspase-dependent [22] and caspase-independent [23] pathways are activated in brain injury after HI. Caspase-3 activation is considered as a reliable marker of the apoptotic neuronal cell death in HI brain injury [24]. Western blot analysis revealed enhanced expression of caspases-3, 8 and 9 in the brain tissues of neonatal rats following HI. The activation of caspases 8 and 9 eventually leads to the activation of caspase-3 [25]. Thus increases in caspase-8 and 9 expression reflects the expression of caspase-3.The increase was in line with the apoptotic cell counts observed, indicating the activation of the caspase cascades that were effectively down-regulated by ellagic acid.
Neuro-inflammation is implicated in hypoxic injuries of neonatal brain [26]. Inflammatory cytokines- TNF-α and IL-1β, along with nitric oxide (NO) produced by activated microglial cells lead to neuronal injury [27].Also targeting MAPK signalling pathways in cerebral injury is valuable in treatment as these pathways are associated with apoptotic signalling as well [28]. Studies have reported that inflammatory mediators produced in cerebral HI injury activate MAPK signalling cascades [29,30]. Our results demonstrate significant activation of JNK, ERK1/2 and p38 suggesting activation of MAPK cascades. Strikingly reduced levels of JNK, p-JNK, p38 and p-38 were observed on ellagic acid pre-treatment, only negligible inhibitory effects were noticed on ERK1/2 and p-ERK1/2. Earlier researches have demonstrated that activated JNK and p38 MAPKs chiefly function to arbitrate cellular stress in cerebral ischemic injury by regulating the activities of intracellular enzymes and transcription factors, associated with cell survival and apoptosis [29]. Thus, suppression of JNK and p-38 MAPK and inflammatory mediators could have possibly contributed to the decrease in neuronal apoptosis.
HI injury resulted in a drastic increase in the expression of NF-κB p65 and p-IκBα. NF-κB regulates genes involved in inflammation and stress responses [31]. Under pathological conditions, IκB is phosphorylated resulting in active NF-κB [32] and gets translocated to the nucleus where it regulates target gene expression. An increase in phosphorylated NF-κB in hypoxic microglia of neonatal rats has been reported previously [33]. Significant increase in the expression levels of IL-6, COX-2, NF-κB and iNOS were also observed. Marked suppression of NF-κB p65, IL -1β, IL-1α and p-IκBα suggests the potent anti-inflammatory efficiency of ellagic acid. Enhanced p65NF-κB expression observed was in line with the expression of TNF-α, COX-2, IL-1α, IL-1β, IL-6 and iNOS following HI. Recent studies reported that the functions of TNF-α and IL-1β were mediated through caspase cascades and iNOS [34]. The down-regulation of inflammatory mediators thus curbs inflammation, suggesting anti-inflammatory effects as one of the mechanisms involved in neuroprotection by ellagic acid.
Conclusion
Ellagic acid reduces brain infarct size and apoptosis of brain cells through mechanisms such as down-regulation of apoptotic pathway proteins, suppression of MAPK cascades and expression of inflammatory mediators. MAPK cascades are critically associated with cell survival and apoptosis. Furthermore, neuro-inflammation adsorbed in HI potentially contributes to apoptosis. Down-regulation of MAPKs and inflammatory protein expression by ellagic acid reveals its protective effects in HI. The results suggest that ellagic acid may be useful in therapeutic strategies for brain injury. However, the potential neuroprotective effects of ellagic acid require further investigation.
Declarations
Acknowledgement
References
Archives


News Updates